
Rubisco purification protocol (sucrose gradient)
Published:
Protocol for the high quality purification of Rubisco, applied to green algae and plants, yielding active, intact Rubisco holoenzyme complexes. This protocol was used for purification of all Rubiscos in a recent Mackinder lab publication.
last updated: 27/08/24
Before starting
Materials
*Essential
- *Ammonium sulphate (e.g.; A4418)
- *DTT (dithiothreitol) (e.g.; R0861)
- *Sucrose
- *~13 mL ultra-centrifuge tubes (e.g.; #344059)
- *Swinging bucket ultra-centrifuge rotor (e.g.; #331362)
- *Anion exchange column/resin (e.g.; #17515901)
- *Superdex 200 gel filtration column (e.g.; #28989335)
- Protein concentration columns (e.g.; Amicon® Ultra-15)
Buffers
Rubisco lysis buffer
- 50 mM Tris-HCl
- 100 mM NaCl
- 0.5 mM EDTA (pH 8.0)
- 5 mM DTT (added fresh)
- Adjust to pH 8.0
10% Sucrose solution
- Rubisco lysis buffer +
- 10% (w/v) sucrose
30% Sucrose solution
- Rubisco lysis buffer +
- 30% (w/v) sucrose
IEX buffer (A)
- 50 mM Tris-HCl
- 50 mM NaCl
- 1 mM DTT
- Adjust to pH 8.0
IEX elution buffer (B)
- 50 mM Tris-HCl
- 1 M NaCl
- 1 mM DTT
- Adjust to pH 8.0
SEC Buffer
- 50 mM Tris-HCl
- 50 mM NaCl
- 5% glycerol (w/v)
- 1 mM DTT
- Adjust to pH 8.0
Essential note
Ensuring that the Rubisco remains in a reducing environment during purification is essential to the integrity of the final preparation and has a great impact on the phase separation propensity and activity of Rubisco. It is recommended that buffers contain 1-5 mM DTT at all times. Otherwise, Rubisco is forgiving and does not require addition of protease inhibitors or special treatment throughout purification. DTT must be added fresh to prevent its degradation.
Protocol
Lysing material
Note 1
This section of the protocol is very variable dependent on the nature of the material to be processed. It is recommended the final lysate is passed through a 0.2 μm filter prior to the next step but extraction techniques should be optimised on a material basis.
- Lyse material in 1x Rubisco lysis buffer (50 mM Tris-HCl, 100 mM NaCl, 0.5 mM EDTA, 5 mM DTT at pH 8.0).
- For green algae (Chlorella/Chlamydomonas), cell disruption by sonication or ‘French press’ has worked well in our hands.
- ~1 g of biomass (wet weight) can be efficiently lysed in ~10 mL of buffer, though excess lysis buffer is preferred, if practical.
- For plant and seaweed material, lysis using a household blender has worked very well in our hands.
- In this case, excess buffer almost always yields better lysis.
- For green algae (Chlorella/Chlamydomonas), cell disruption by sonication or ‘French press’ has worked well in our hands.
- Centrifuge lysate at ~50,000 g for 30 minutes at 4 °C.
- Filter supernatant through 0.2 µm membrane.
- At this point, the solution should be green, but free of large thylakoid fragments. Smaller thylakoid fragments will be removed in the following steps.
- If it is the first time performing an extraction on the specified material, here is a good place to take a sample for a gel to estimate your lysis efficiency.
Ammonium sulphate precipitation
Note 2
We have found ammonium sulphate precipitation very useful for removing smaller thylakoid fragments and crucially, contaminating chlorophyll-binding proteins (e.g. LHCBs), that will bind anion exchange media and reduce the efficiency and purity of subsequent purification steps. The precipitation is completed in two steps; one to remove unwanted large proteins and thylakoid proteins and a second to precipitate the remaining soluble proteins (including Rubisco). In short; discard the first pellet and keep the second!
- Dilute the lysate from the previous step ~2-fold in Rubisco lysis buffer and measure the volume in a measuring cylinder.
- Ammonium sulphate in larger volumes is much more controllable and in turn appears to be more efficient.
- Whilst the lysate is stirring, add ammonium sulphate powder to a final concentration of 190 mg mL-1 and stir for 1-2 hours at 4 °C.
- Centrifuge at ~50,000 g for 10 minutes at 4 °C.
At this stage, dark green pellets should be observed, which contain thylakoid material and unwanted large proteins. The supernatant should be largely free of green pigment, and have a yellow-ish colouration.

- Decant the supernatant and, whilst stirring, add ammonium sulphate powder to a final concentration of 390 mg mL-1 (a further 200 mg mL-1 to previous step) and stir for 1-2 hours at 4 °C.
- Centrifuge at ~50,000 g for 10 minutes at 4 °C.
- Very light green/brown pellets should be observed and contain most of the Rubisco.
- Resuspend the pelleted fraction in IEX buffer (50 mM Tris-HCl, 50 mM NaCl, 1 mM DTT at pH 8.0).
- The objective here is to resuspend the pelleted material in the smallest volume in which all of the protein is soluble. This is crucial for the sucrose gradient centrifugation to work correctly.
- As a rough guide, the pellets from 3 L of Chlamydomonas/Chlorella can usually be resuspended to 6 mL.
- To check if the protein is sufficiently solubilised, attempt to float a small volume on top of 10 % (w/v) sucrose solution. The protein solution should sit on top of the sucrose and not sink into it. If it does so, resuspend in a larger volume. Alternatively, centrifuge a small volume at ~20,000 g and check for a pellet (bad).
Sucrose gradient ultra-centrifugation
Note 3
Sucrose gradient centrifugation works on the principle of differential mobility of protein species through dense sucrose solutions dependent on their molecular weight and hydrodynamic radius. As Rubisco is a relatively large protein (~550 kDa), it is particularly well-suited to separation from other, smaller soluble proteins using this technique. In this protocol we use a linear 10-30 % (w/v) gradient. Though gradients can be formed manually (see below), it is much more reproducible to use automated approaches to do so.
- Pour linear 10-30 % (w/v) sucrose gradients from solutions formed in IEX buffer.
- We now routinely use a gradient master to form gradients using the two base solutions (10 % and 30 % (w/v)) as per manufacturer instructions instructions. We use the ‘long’ caps, which leave an approximate volume of 1 mL on top of the gradients for sample loading.
- Gradients can be poured manually without specialist equipment as per below:
- Using a long syringe needle layer ~4 mL of 10 % (w/v) sucrose solution into the bottom of each of the ultracentrifuge tubes.
- By inserting the syringe needle to the bottom of the tube, underlay ~4 mL of 20 % (w/v) sucrose solution.
- Underlay ~4 mL of 30 % (w/v) sucrose solution.
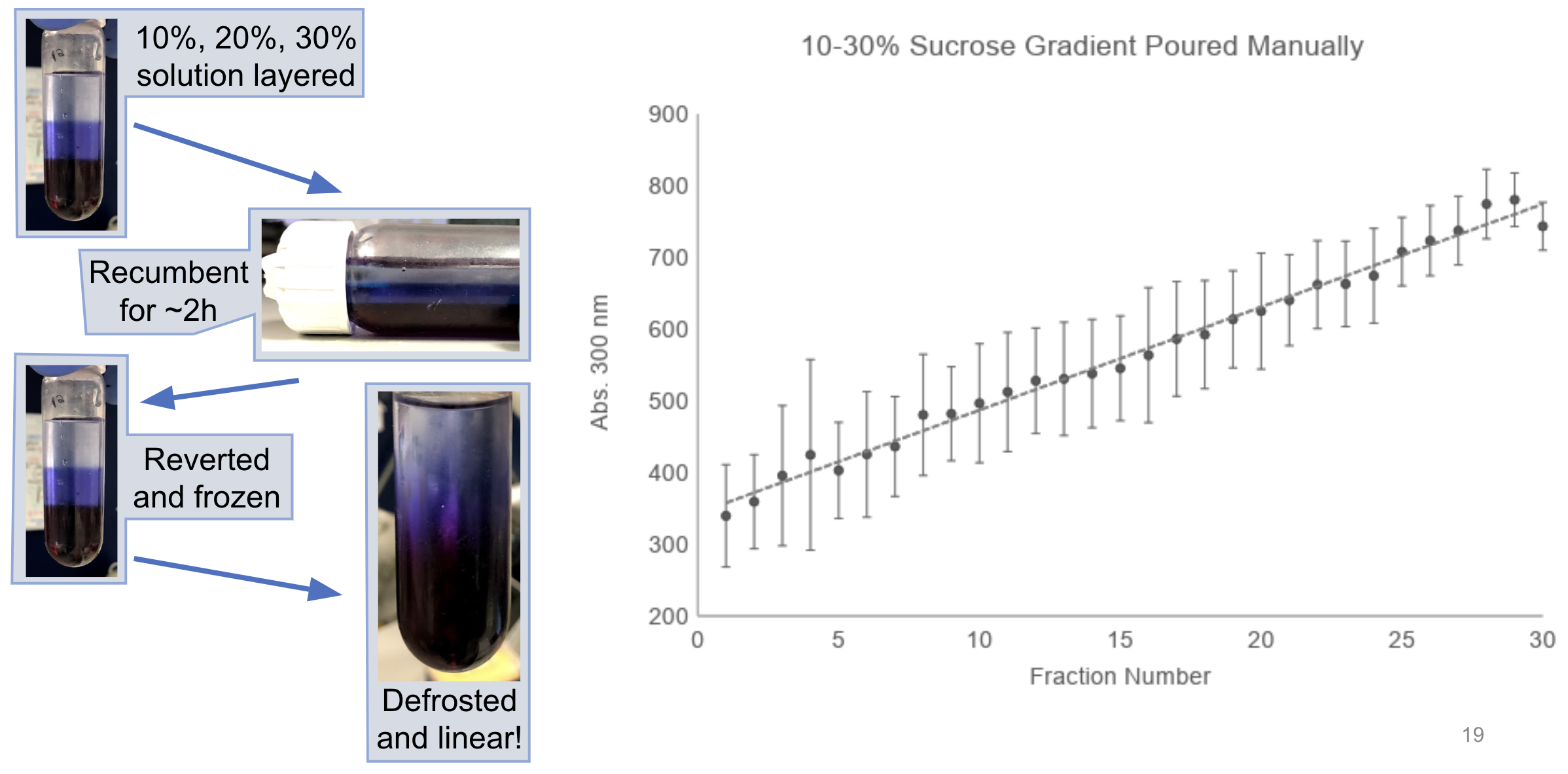
- Cover the top of the tube with parafilm and slowly lay the tubes flat such that each of the layers is spread across the length of the tube.
- Leave the layers to linearise by diffusion for ~2 hours at room temperature.
- Freeze the gradients and thaw when ready to use.
The combination of diffusion and freeze/thaw should allow linearisation of the gradients (shown here with a dye for illustration):
- Load the resupsended protein solution from the ammonium sulphate precipitation onto the gradients.
- Centrifuge in a swinging bucket rotor (e.g. SW41-Ti) at 37,000 rpm for 16.5 hours at 4 °C.
- Fractionate one of the gradients into 500 µL fractions by pipetting from the top of the gradient down, ensuring the tip is always taking from the very top of the solution.
- At this point, we often run a quick diagnostic gel to check which fractions the Rubisco is distributed in as there is often small differences between different preparations.
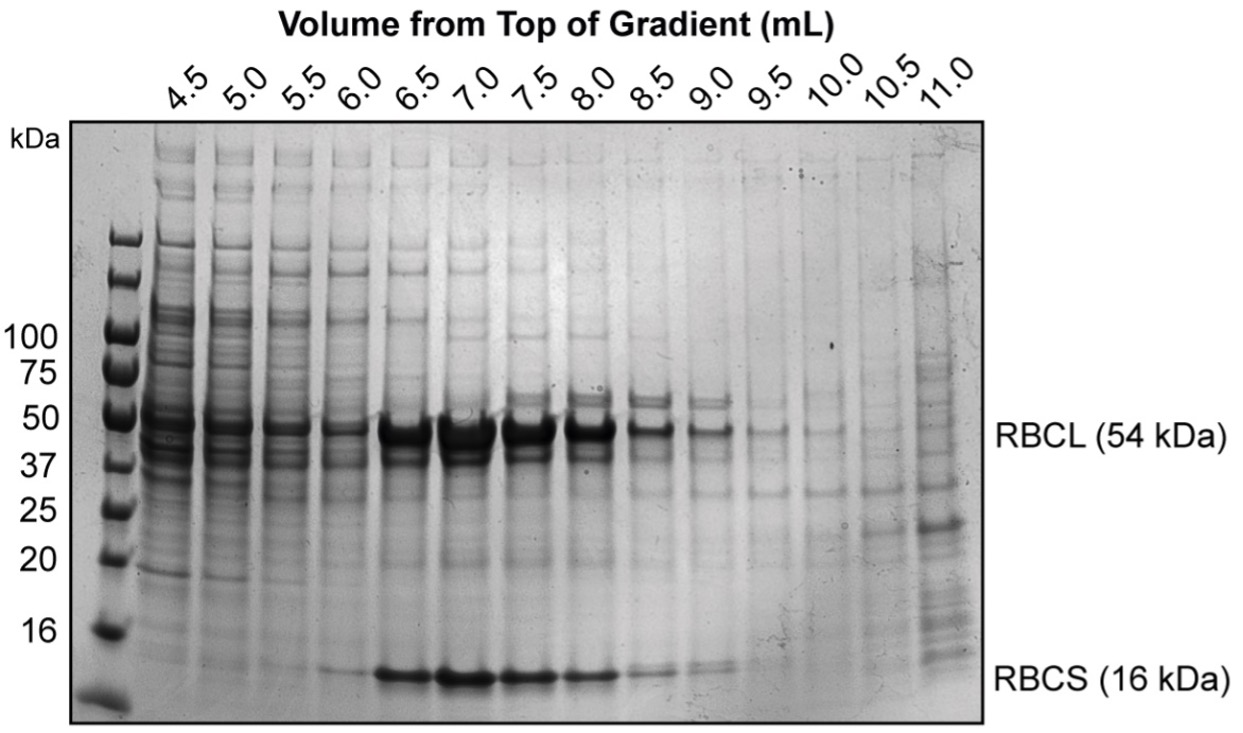
- At this point, we often run a quick diagnostic gel to check which fractions the Rubisco is distributed in as there is often small differences between different preparations.
- Pool the Rubisco-containing fractions from each of the gradients (6.5 - 9.0 mL from above image).
Anion exchange
Note 4
We use cheap HiTrap Q XL 5 mL anion exchange columns for our ion exchange procedures as we are often completing large preparations that will slowly accumulate small amounts of chlorophyll and other pigments on the columns over time. Higher performance columns can be used in place here if desired.
- Dilute the pooled Rubisco fraction two-fold in IEX buffer (A).
- Though not strictly necessary, we find this improves the retention of Rubisco on the IEX column.
- Pass the Rubisco fraction over an anion exchange column equilibrated with IEX buffer (A).
- Wash the column back to ~0 mAU with IEX buffer (A).
- Elute the bound Rubisco with a linear gradient to 100% IEX elution buffer (B) (50 mM Tris-HCl, 1 M NaCl, 1 mM DTT at pH 8.0) over 10-20 column volumes.
- Rubisco should elute between 250 mM and 300 mM NaCl:
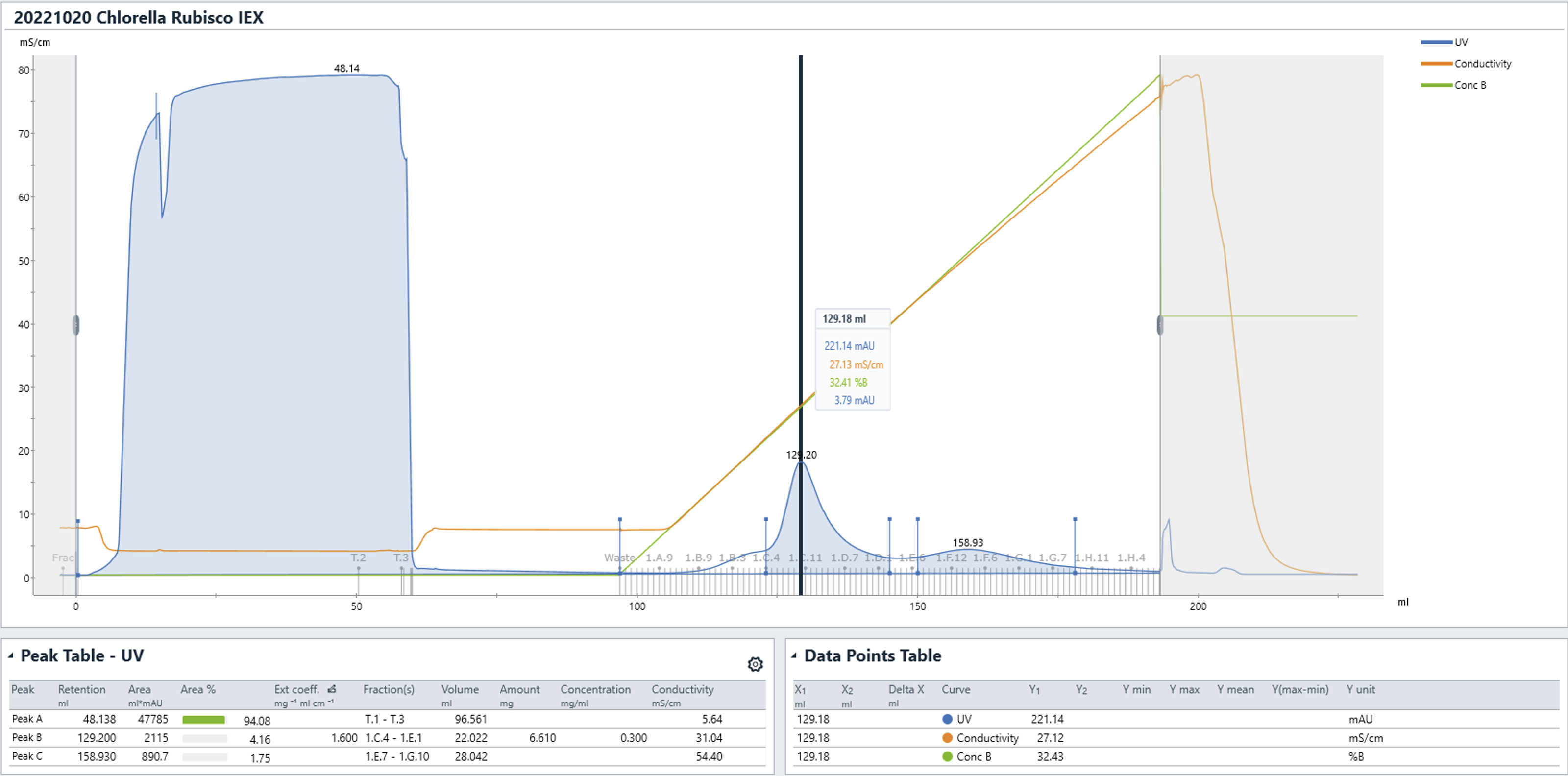
- Rubisco should elute between 250 mM and 300 mM NaCl:
- Pool Rubisco fractions and concentrate (using e.g.</a>).
Size exclusion chromatography
Note 5
Size exclusion chromatography (SEC) should only be used as a final ‘polishing’ step. If your fractions still contain significant impurities, pigments, or nucleic acids, performing SEC will not yield significant improvements in purity. Repeating anion exchange steps with higher performance (e.g. SOURCE 15Q) columns can somewhat overcome poor purity preps at this stage.
- Equilibrate column with SEC buffer (50 mM Tris-HCl, 50 mM NaCl, 5% glycerol (w/v), 1 mM DTT at pH 8.0) and load Rubisco sample.
- We routinely use columns from the Superdex 200 preparative grade (pg) or increase families - typically 16/600 pg or 10/300 increase.
- The Rubisco holoenzyme elutes from a 16/600 column at ~55-65 mL and ~9-11 mL on a 10/300 GL column.
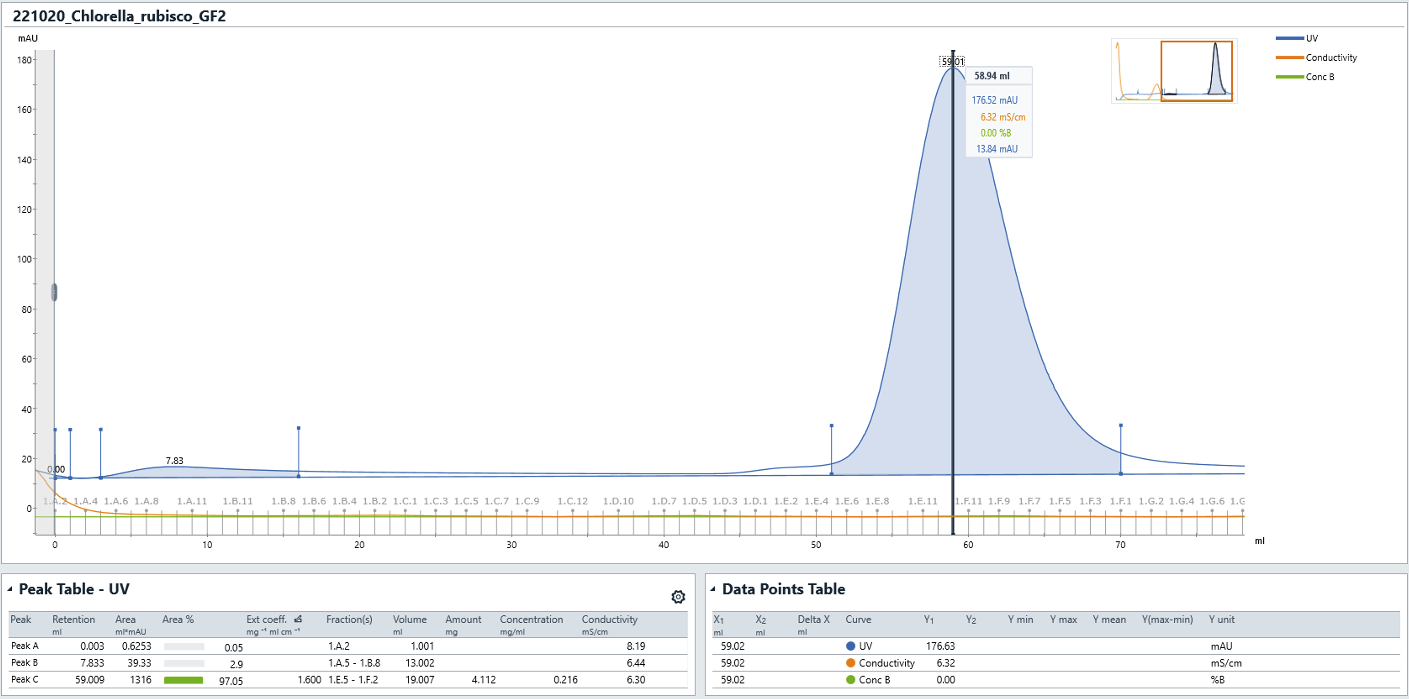
- Pool and concentrate the Rubisco fractions.
- The fraction can be flash-frozen in liquid nitrogen and stored at -70 °C in SEC buffer.
N.B.</br> Without reducing agents, we have observed the small subunits of the holoenzyme to fall off, leaving an L8 octamer and free small subunits in solution. This can be hard to detect without a known ‘good’ sample run adjacent on a native PAGE gel.